| [Related articles/posters: 033 003 028 ] |


Modelling SN2
nucleophilic substitution at silicon by structural correlation with X-ray
crystallography.
Alan R.
Bassindale, David J. Parker, Peter G. Taylor.
Department of Chemistry, Open University, Walton Hall, Milton Keynes,
MK7 6AA, United Kingdom.
Norbert Auner, Bernhard Herrschaft.
Institut für
Chemie der Humboldt-Universität
zu Berlin, Fachinstitut für Anorganische und Allgemeine Chemie,
Hessische Str. 1-2, D-10115 Berlin, Germany.
The trajectory of bimolecular nucleophilic substitution
at silicon is modelled using a series of closely related five-coordinate
silicon complexes based on quinoline ligands. Each complex contains an
internal 'nucleophile' which undergoes an intramolecular interaction with
the silicon. By careful choice of 'leaving group' at the silicon, different
extents of 'substitution' are observed. The extent of bond formation in
solution and in the solid state are measured using multi-nuclear NMR spectroscopy
and single crystal X-ray crystallography.
Introduction
Modelling the trajectory of an SN2 reaction pathway was first performed by Dunitz over 15 years ago.1
The technique known as structural correlation involves the collection of X-ray crystallographic data for compounds containing the molecule of interest and then sequencing the structures on the basis of key crystal data (usually inter-atom distances).
The structures are arranged
so that a gradual deformation is observed, each structure representing
a frozen snapshot of the substitution at a particular point on the
modelled reaction profile.
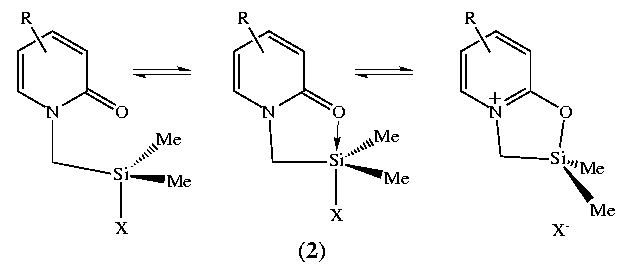
Pestunovich
et al 2 were the first to apply
this methodology to substitution at silicon in their study of N-(amidomethyl)-chlorosilanes
(1).
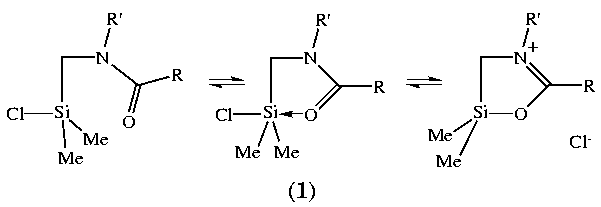
In response, a method for modelling the substitution in solution was developed by Bassindale3 using 13C and 29Si NMR spectroscopy as a monitoring tool. The system first studied was that using substituted 2-pyridone ligands (2) with a variety of ring substituents and leaving groups.
By varying R and X in
a series of 25 such complexes, the substitution was mapped. We have since
extended this technique to a series of 2-quinolinone
analogues (3).4
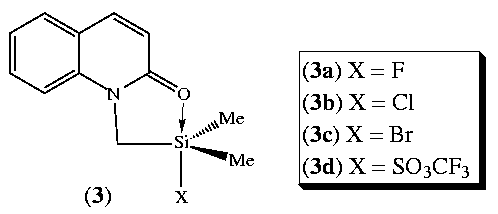
We present here the results of our X-ray crystallographic
study of complexes (3a-d) and so for the first time compare solution and
solid state descriptions of the complexes and the substitution they model.
Structure of fluoride complex (3a)
|
Si-F = 1.68Å |
|
Si-O = 2.06Å |
|
N-C9-Si = 113.3o |
|
Si-O (typical) = 1.50Å |
|
Si-F (typical) = 1.56Å |
Fluoride is the poorest leaving group of the series and so
models the earliest point in the substitution with the longest nucleophile-silicon
distance; the amido oxygen being the nucleophile and the halogen the leaving
group.
Structure
of chloride complex (3b)
|
Si-Cl = 2.32Å |
|
Si-O = 1.94Å |
|
N-C9-Si = 109.0o |
|
Si-Cl (typical) = 2.03Å |
With an improved leaving group the oxygen is closer to
the silicon by 0.12Å. The Si-Cl bond is markedly stretched from its
normal length. The N-C9-Si angle decreases slightly as ring
closure begins to take place.
Structure
of bromide complex (3c)
|
Si-Br = 2.65Å |
|
Si-O = 1.85Å |
|
N-C9-Si = 106.5o |
|
Si-Br (typical) = 2.17Å |
With the nucleophile a further 0.09Å closer to the silicon, the Si-X bond is considerably stretched and weakened as the strengthened nucleophilic interaction causes it to depart further. The N-C9-Si angle continues to decrease.
Structure of triflate complex (3d)
|
Si-O(triflate) = 2.76Å |
|
Si-O(carbonyl) = 1.74Å |
|
N-C9-Si = 103.7o |
|
Si-O (typical) = 1.50Å |
The nucleophile is now 0.11Å closer to the silicon
and the substitution is close to completion. The interaction of the triflate
group with the silicon is best described as electrostatic. There are no
further silicon-oxygen distances in the crystal corresponding to further
bonding interaction.The axial bond angles are somewhat distorted from the
180° ideal due to the intramolecular nature of the substitution.
To describe the degree to the which the Si-X bond is "stretched"
by the incoming nucleophile, a percentage Si-X extension term is
introduced. We define 0% extension as that of a typical Si-X bond and 100%
extension as the sum of the crystal ionic radii for Si and X.
The key solid-state data,
along with the results of the NMR solution work are presented in the following
table. ORTEP diagrams and further crystal data are summarised here.
|
Complex |
|
|
|
|
|
X |
F |
Cl |
Br |
SO3CF3 |
|
Si-X /Å |
1.68 |
2.32 |
2.65 |
2.76 |
|
% Si-X extension* |
29 |
67 |
109 |
126 |
|
Si-Odonor /Å |
2.06 |
1.94 |
1.85 |
1.74 |
|
% Si-Odonor (solution studies)4 |
35 |
63 |
78 |
93 |
![]()
The geometry at the silicon remains trigonal-bipyramidal
for the majority of the 'reaction'. This is particularly suprising for
complex (3a) where the nucleophile-silicon interaction is particularly
weak and the silicon is essentially tetracoordinate.
The relationship between the extent of Si-O bond formation
and the degree of Si-X bond stretching is shown below.
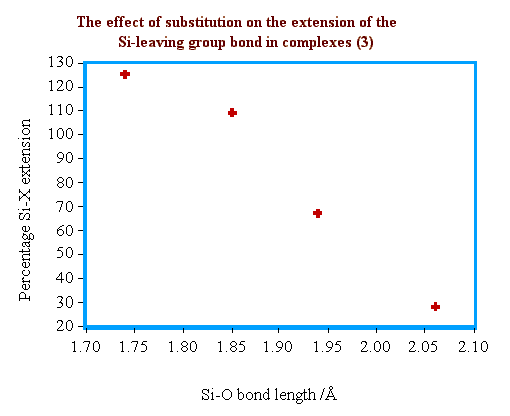
As expected, the degree of Si-X bond stretching increases with strengthening nucleophilic attack. In the case of the triflate structure (3d) the stretching is great enough that the Si-X distance is more characteristic of an inter-ion contact distance than a covalent interaction.
We can also compare the extent of Si-O bond formation between solid state and solution.
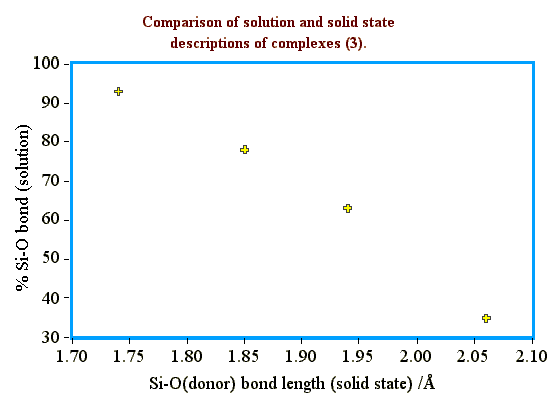
These studies show an excellent correlation between the solution
results and X-ray data.
We have successfully modelled the nucleophilic SN2
substitution at silicon both in solution and in the solid state for a series
of complexes (3) based on 2-quinolinone ligands and shown how the
geometry of the complexes is very similar between the solid state and solution
phases.
From the crystallographic data we have demonstrated the
concerted Si-nucleophile bond shortening and Si-leaving-group extension
that is characteristic of this mechanism.
1. D. Britton,
J.D. Dunitz; J. Amer. Chem. Soc.; 1981, 103, 2971-2979.
2. V.F.
Sidorkin, V.V. Vladimirov, M.G. Voronkov, V.A. Pestunovich; J. Mol.
Struct. (Theochem); 1991, 228, 1-9.
3. (a) A.R. Bassindale, M. Borbaruah; J. Chem. Soc. Chem. Commun.; 1991, 1499-1501. (b) A.R. Bassindale, M. Borbaruah; J. Chem. Soc. Chem. Commun.; 1991, 1501-1503.
4. A.R.
Bassindale, D.J. Parker, P.G. Taylor; unpublished results.